Step 3
Step 3a: Calculating the charge for a list of many peptides
Using the pep_charge() function, you were able to calculate the charge for 5 peptides at 3 pH values each. That involved using the function 15 times, which is doable, but slightly tedious. You could have done this same exercise with any of the online tools referenced previously, and although it might not have been as educational, you would have achieved the same result. Now say that you have a mixture of 50, or 100, or even 1000 peptides. It suddenly becomes incredibly time-consuming to do all these calculations one at a time. Luckily, because you have the pep_charge() function available in a versatile coding language, you are able to automate some of these calculations.
Exercise (3a)
Here is a list of 52 peptides that you have identified in your sample after a proteomics experiment:
HSTIFENLANK, HTFMGVVSLGSPSGEVSHPR, HTLNQIDEVK, IDVHLVPDR, IRLENEIQTYR, ITQVLHFTK, IVSSAMEPDREYHFGQAVR, KAEEEHLGILGPQLHADVGDK, KCPLPGTAAFK, KPVDEYKDCHLAQVPSHTVVAR, KPVEEYANCHLAR, KQTALVELVK, KVPQVSTPTLVEVSR, KYLYEIAR, LDELRDEGK, LKYENEVALR, LQHLENELTHDIITK, LSINTHPSQKPLSITVR, LVHVEEPHTETVR, LVNEVTEFAK, LVRPEVDVMCTAFHDNEETFLK, LVRPEVDVMCTAFHDNEETFLKK, LYHSEAFTVNFGDTEEAKK, MRPSTDTITVMVENSHGLR, NECFLQHKDDNPNLPR, PDYSVVLLLR, PSLVEQVFLDK, PVPGHVTVSICR, QEPERNECFLQHK, QEPERNECFLQHKDDNPNLPR, QHIEAIDVR, QNCELFEQLGEYK, QSVEADINGLRR, QEPERNECFLQHKDDNPNLPR, QTALVELVK, RHPDYSVVLLLR, RPCFSALEVDETYVPK, RYLYEIAR, SLHTLFGDK, SPAFTDLHLR, SSNLIILEEHLK, TFHADICTLSEK, THLAPYSDELR, THLPEVFLSK, VDLSFSPSQSLPASHAHLR, VFDEFKPLVEEPQNLIK, VHTECCHGDLLECADDR, VHTECCHGDLLECADDRADLAK, VLANGHAPYSR, VLGYVVEMQPK, VPQVSTPTLVEVSR, VQFELHYQEVK
And you want to know the charge of all of them at pH 7.
The code below imports them into R, storing them in a list called peptides. Copy and paste, and then run this in your R console.
peptides <- list("HSTIFENLANK", "HTFMGVVSLGSPSGEVSHPR", "HTLNQIDEVK", "IDVHLVPDR", "IRLENEIQTYR", "ITQVLHFTK", "IVSSAMEPDREYHFGQAVR", "KAEEEHLGILGPQLHADVGDK", "KCPLPGTAAFK", "KPVDEYKDCHLAQVPSHTVVAR", "KPVEEYANCHLAR", "KQTALVELVK", "KVPQVSTPTLVEVSR", "KYLYEIAR", "LDELRDEGK", "LKYENEVALR", "LQHLENELTHDIITK", "LSINTHPSQKPLSITVR", "LVHVEEPHTETVR", "LVNEVTEFAK", "LVRPEVDVMCTAFHDNEETFLK", "LVRPEVDVMCTAFHDNEETFLKK", "LYHSEAFTVNFGDTEEAKK", "MRPSTDTITVMVENSHGLR", "NECFLQHKDDNPNLPR", "PDYSVVLLLR", "PSLVEQVFLDK", "PVPGHVTVSICR", "QEPERNECFLQHK", "QEPERNECFLQHKDDNPNLPR", "QHIEAIDVR", "QNCELFEQLGEYK", "QSVEADINGLRR", "QEPERNECFLQHKDDNPNLPR", "QTALVELVK", "RHPDYSVVLLLR", "RPCFSALEVDETYVPK", "RYLYEIAR", "SLHTLFGDK", "SPAFTDLHLR", "SSNLIILEEHLK", "TFHADICTLSEK", "THLAPYSDELR", "THLPEVFLSK", "VDLSFSPSQSLPASHAHLR", "VFDEFKPLVEEPQNLIK", "VHTECCHGDLLECADDR", "VHTECCHGDLLECADDRADLAK", "VLANGHAPYSR", "VLGYVVEMQPK", "VPQVSTPTLVEVSR", "VQFELHYQEVK")You may not know exactly how to make your function run individually on many different peptides, but luckily you found another published script that does something related. In this case, the function pep_length() calculates the number of amino acids in a sequence. Then, below that, the author wrote a for loop that goes through a list of peptides (just like the one you have!) and calculates and prints the length for each one. Copy and paste the code below to your ‘.R’ file, and run it.
#function that returns the number of amino acids in a peptide
pep_length <- function(peptide) {
length <- nchar(peptide)
return(length)
}
#loop through a list of peptides and print out the peptide followed by the number of amino acids
for (pep in peptides) {
#print out the peptides, one at a time. 'pep' is the variable that is a different peptide each loop
print(pep)
#calculate the length of the peptide
peptide_length <- pep_length(peptide = pep)
#print out the peptide length
print(peptide_length)
}If you ran the peptides list command earlier and this new block of code, you should see an output that printed each peptide followed by the number of amino acids it contains. Here are what the first three printouts should look like:
## [1] "HSTIFENLANK"
## [1] 11
## [1] "HTFMGVVSLGSPSGEVSHPR"
## [1] 20
## [1] "HTLNQIDEVK"
## [1] 10Now you want to do essentially the same thing, but instead of printing out the length, print out the charge at pH 7. How can you modify the code to print out the charge? Note that you don’t have to change anything in the pep_length function (The first 4 lines of the above code). The changes will occur in the for loop that starts for (pep in peptides) {…
You may already have an idea on how to make this happen, that’s great! However, let’s go through an exercise to logically think through what needs to be changed. Here are three general questions we can ask, that sometimes help to figure out a good next step.
What does the code do? What are the inputs and what are the outputs?Click for a possible answer
We are talking about thefor loop here. The for loop takes in a list of peptides, and in this case the list is called peptides. For each item in the list, it prints out the peptide (within the for loop, the variable pep refers to whichever peptide the loop is currently on). The loop calculates the length of the peptide using the function pep_length(), and stores that value in the variable peptide_length. Then the for loop prints out the variable peptide_length, which is a number that was calculated in the previous line. The process is repeated for each peptide in the list peptides
Click for a possible answer
We have a list of peptides, also calledpeptides. We want to use this as the input. For each peptide, we want to calculate the charge at pH 7.0. The outputs will be a printout of the peptide, followed by a printout of its charge
Click for a possible answer
The input to the for loop is exactly the same: a list of peptides called peptides. Great! The existing loop prints out the name of each peptide (pep in this case). That’s also fine to stay as is.
The existing loop calculates a length, and saves it as a variable peptide_length. We don’t want that. However, you do know a function (from exercises 1 and 2) that calculates the charge of a peptide, which is what we want! How about we use that function instead of pep_length(). Instead of saving the charge value to the variable peptide_length, how about we save it to a different new variable. Maybe peptide_charge will work? You can copy the formatting from the existing for loop, but change the function and the name of the variable. The function pep_charge() takes two arguments: a peptide, and a pH. The peptide part is the same as the existing loop, but we need to specify the pH. Look back at exercise 1 to remember how to format the `pep_charge() function.
peptide_length. We just made a new variable called peptide_charge. Can you print that out instead?
Hopefully answering those questions yourself, or reading through a possible answer above helped you think through the changes that you need to make. Copy and paste a screenshot of your modified code that prints out the charges of all 52 peptides into your assignment (You don’t need to take a screenshot of all the values, just a picture of the for loop that you modified. Remember that when you change the code, make sure you highlight it and click ‘Run’ again.
If it worked correctly, you might get an output that includes the correct charges, but also includes the dataframe used to calculate the charge for each peptide. That is a lot of information, and not necessary in this case. Go back to your original pep_charge() function from exercise 1. Find the line that prints out the net_charge_df and delete it or comment it out (putting a # symbol in front of a line of code tells the computer to ignore it - this is referred to as ‘commenting out a line’). Re-run that original pep_charge() function to save the change, and then re-run your ‘for loop’ to print out the values. Is that more manageable?
Click for an additional hint if you’re stuck
One very important ‘motif’ in R coding is seen in the original for loop above. Lets take a closer look at this particular line:
peptide_length <- pep_length(peptide = pep)peptide_length is a variable, pep_length() is a function, and peptide = pep within the parenthesis is the argument for the function. <- is the assignment symbol in R (think of it as an arrow pointing to the left). It means that the value on the right of the symbol is stored in the variable written to the left of the symbol.
A more general version of this line of code is:
variable <- function(argument1 = input_value1, argument2 = input_value2)The function does some sort of calculation based on the inputs, and returns a value. This value is then stored in the variable.
Can you copy the line of code above, but replace each section with the information needed to calculate the charge on each peptide within thefor loop? The variable is a new one: peptide_charge. The function is from exercise 1: pep_charge(). argument1 for this function is called peptide, and it is = to pep within the for loop. argument2 is called pH, and it is = to 7.0 for the purpose of this calculation. Remember to re-highlight and press ‘Run’ when you’ve changed your loop!
Step 3b: Predicting m/z values for peptides in MALDI experiments
Matrix Assisted Laser Desorption Ionization Time of Flight (MALDI-TOF) mass spectrometry is an excellent way to quickly obtain mass values for analytes in a solution. In this technique, the sample is combined with a ‘matrix’ (usually some sort of organic acid). This matrix-sample combination is dried on a plate, and placed in the instrument. A laser excites the sample and desorbs both matrix and sample from the plate. In this high energy cloud, some analyte obtains an extra proton from the matrix and becomes positively charged. The m/z value of this analyte is measured by the time it takes to reach an ion detector, after being accelerated by an applied voltage. Analytes with smaller m/z values will travel faster than those with larger m/z values.
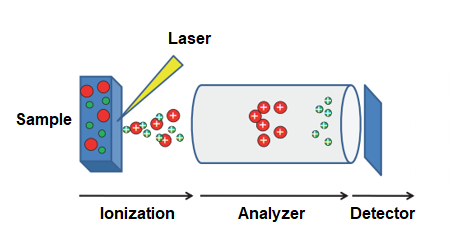
MALDI-TOF Schematic (Wikipedia)
Exercise (3b)
You run each of the fractions from exercise 2 on a MALDI-TOF instrument, and obtain mass spectra, which show signal at specific m/z values. You want to calculate the expected m/z values for each of your peptides to see if they are present. Luckily, you find 2 more helpful published R functions. The first takes a peptide, and returns the mass of the peptide in Daltons. The second takes a mass and a charge value and returns m/z. Copy and paste both of these functions into R and run them.
#calculate peptide mass
#create_dictionary. Each one letter code corresponds to a mass value. Must be run before the function
one_letter <- c('A','R','N','D','C','E','Q','G','H','I','L','K','M','F','P','S','T','W','Y','V')
masses <- list(71.03711381, 156.1011111, 114.0429275, 115.0269431, 103.0091845, 129.0425931, 128.0585775, 57.02146374, 137.0589119, 113.084064, 113.084064, 128.0949631, 131.0404846, 147.0684139, 97.05276388, 87.03202844, 101.0476785, 186.079313, 163.0633286, 99.06841395)
names(masses) <- one_letter
#the function to calculate the mass
pep_mass <- function(peptide){
#convert the string to uppercase
peptide <- toupper(peptide)
#split the peptide into individual letters. [[1]] is required to select the individual values as opposed to a list
splitpep <- strsplit(peptide,'')[[1]]
#create a variable to store the mass. Set it to 0 to start
total_mass <- 0.0
#loop through each residue ('AA' in this case) in the peptide
#first ensure that it is within the list of one letter codes above (by checking that it isn't null)
#then add the corresponding mass for that one letter code to the total mass. masses[[AA]] accesses the mass corresponding to the specific amino acid
for (AA in splitpep) {
if (!is.null(masses[[AA]])) {
total_mass <- total_mass + masses[[AA]]
}
}
#add the mass of H2O (for the n and c terminus... the masses used assume peptide bonds between amino acids)
total_mass <- total_mass + 18.010565
#return the total mass
return(total_mass)
}
#A function that calculates m/z given a peptide and a pH
m_over_z <- function(peptide, pH = 7) {
peptide_mass <- pep_mass(peptide)
peptide_charge <- pep_charge(peptide, pH = 7)
mass_charge_ratio <- peptide_mass / peptide_charge
return(mass_charge_ratio)
}Lets try it on one of our favorite peptides, DIAK. You should run this in your own console as well.
m_over_z("DIAK", pH = 7)## [1] -18971.03Hmmm that output seems very high (and negative). What could be wrong? Lets try calculating charge and mass separately.
pep_charge("DIAK", pH = 7)## [1] -0.02347019pep_mass("DIAK")## [1] 445.2536The mass looks reasonable, but the charge is very close to zero (we actually knew this already from exercise 1). It turns out, when we are dividing mass / charge, we are dividing by a number that is essentially zero, causing the m/z value to skyrocket.
If you think about MALDI however, we don’t actually know what the pH is, and so assuming it to be 7 is probably not accurate. In addition, based on your extensive knowledge of MALDI with peptides, you remember that almost all peptides end up with a +1 charge (due to one proton) when they are ionized with this technique. How could you modify the m_over_z() function to make it applicable to MALDI? The important aspects are:
- The charge will always be +1
- The mass will be the mass of the peptide plus the mass of a proton which is approximately 1.008 Da.
Note that you don’t have to make changes in the pep_mass function which is the first chunk of code above. Instead, the changes will take place within the m_over_z function section, which is the last 6 lines.
Lets think through those same three questions we asked in exercise 3a. Write out answers to these questions in your assignment.
What does the code do? What are the inputs and what are the outputs?
What do we want the code to do? What inputs do we have, and what do we expect as outputs?
What parts of the existing code already do what we want, and what parts need to change?
Click for an additional hint if you’re stuck
To calculate m/z we need to have a mass divided by a charge. Can you see where this is done in the original function? Which variable stores mass, and which stores charge? Do we need the charge varibale, or is that a fixed value for MALDI?
Dividing in R is as simple as taking one variable, writing ‘/’ and writing another variable. For example, if I had a varible tacos that was the number of tacos I bought, and another variable cost that was how much I paid for all the tacos, I could calculate a variable price_per_taco by doing:
price_per_taco <- tacos / costThe same goes for adding. If I left a tip of $2 when I bought the tacos, I could calculate a variable total_cost by doing:
total_cost <- cost + 2Note that the pep_mass() function above is designed for calculating mono-isotopic mass. We know that there are different isotopes that result in shifted masses, but for this purpose we are interested in the mono-isotopic mass because that is generally the most intense peak in peptide MALDI spectra.
Take a screenshot of your modified m_over_z() function and paste it in your assignment. What are the expected MALDI m/z values for each peptide from Exercise 2?